(T1430-11-60) Determining Impact of Particle Size and Crystal Quality on Formation of Nitrosamine in API and Drug Formulations
Purpose: The purpose of this project is to develop an understanding of the role of solid-state reactive species (SSRS) in the formation of nitrosamines, and to describe methodologies that can be used to both determine their concentration and to mitigate their formation.
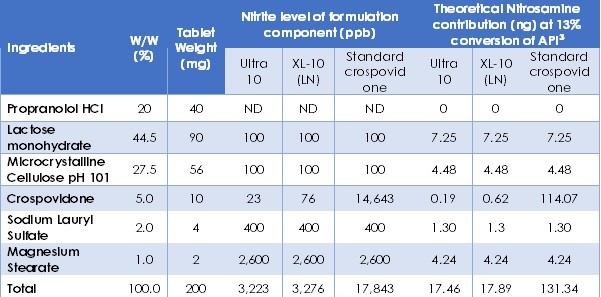
Methods: The SSRS within several model compounds including 4-phenylpiperdine, ranitidine hydrochloride, were created with different manufacturing methods. Samples were cryo-milled or ball-milled for different length. The amount of SSRS was quantified with Solid-state NMR 1H T1 relaxation time (T1). Both milled and bulk samples were stored in different accelerated conditions. Ranitidine hydrochloride would degrade to N-nitrosodimethylamine (NDMA) and 4-phenylpiperdine would degrade to N-nitroso-4-phenylpiperdine (NiPP) at presence of nitrite. The levels of degradation product were measured by LC-MS/MS or LCMS at multiple time points. The surface area of milled sample was measured by Brunauer-Emmett-Teller (BET) surface area analyzer. After understanding the impact of SSRS in drug substance degradation process, this research will then focus on stability of drug substance/drug formulations under the influence of SSRS. The same model compounds will be tested with different formulations, primarily differing in the total nitrite content in the formulations. Total nitrite content is the limiting factor in the nitrosamine formation. The effect of SSRS and nitrite content on the degradation rate will be both evaluated.
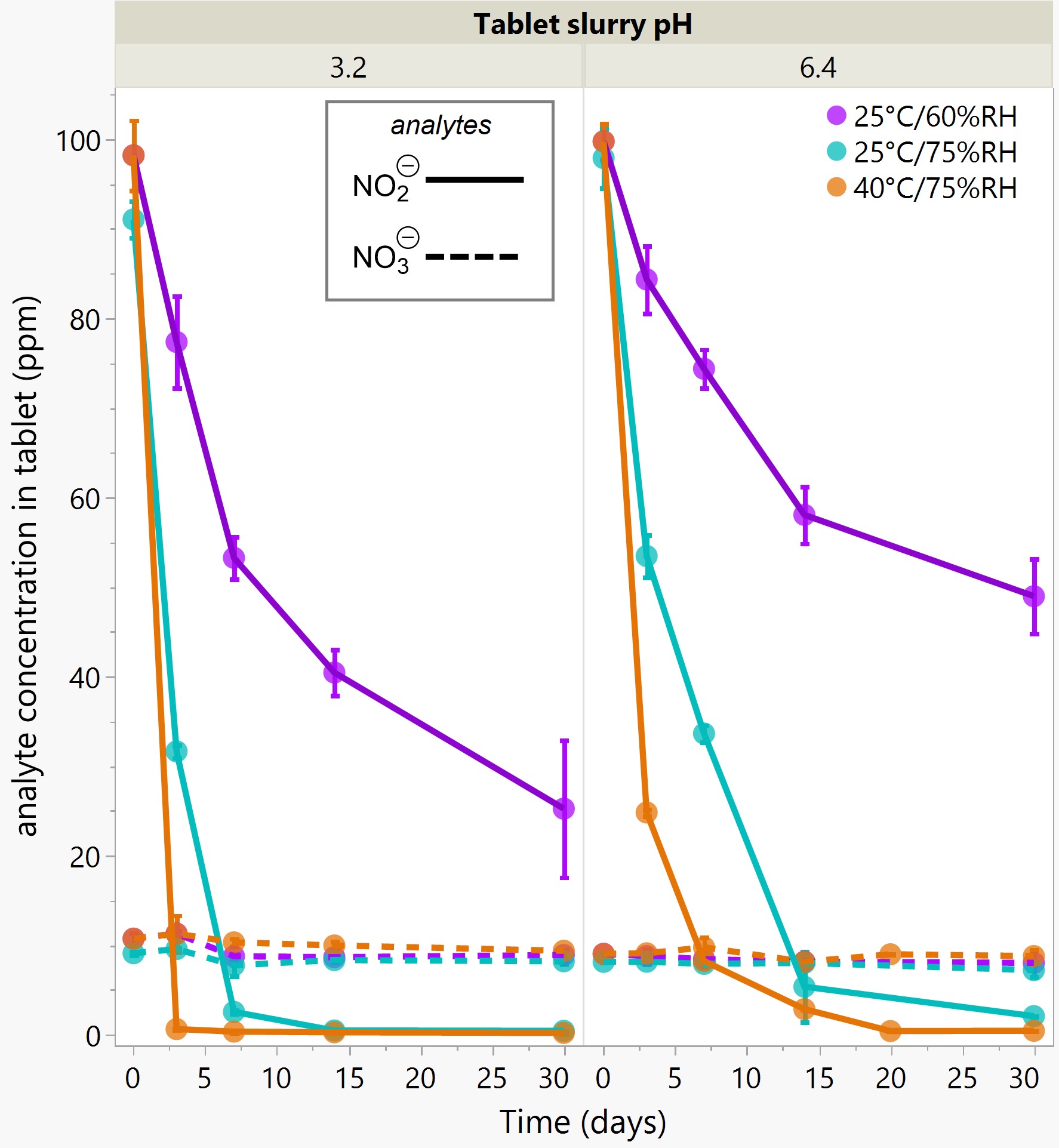
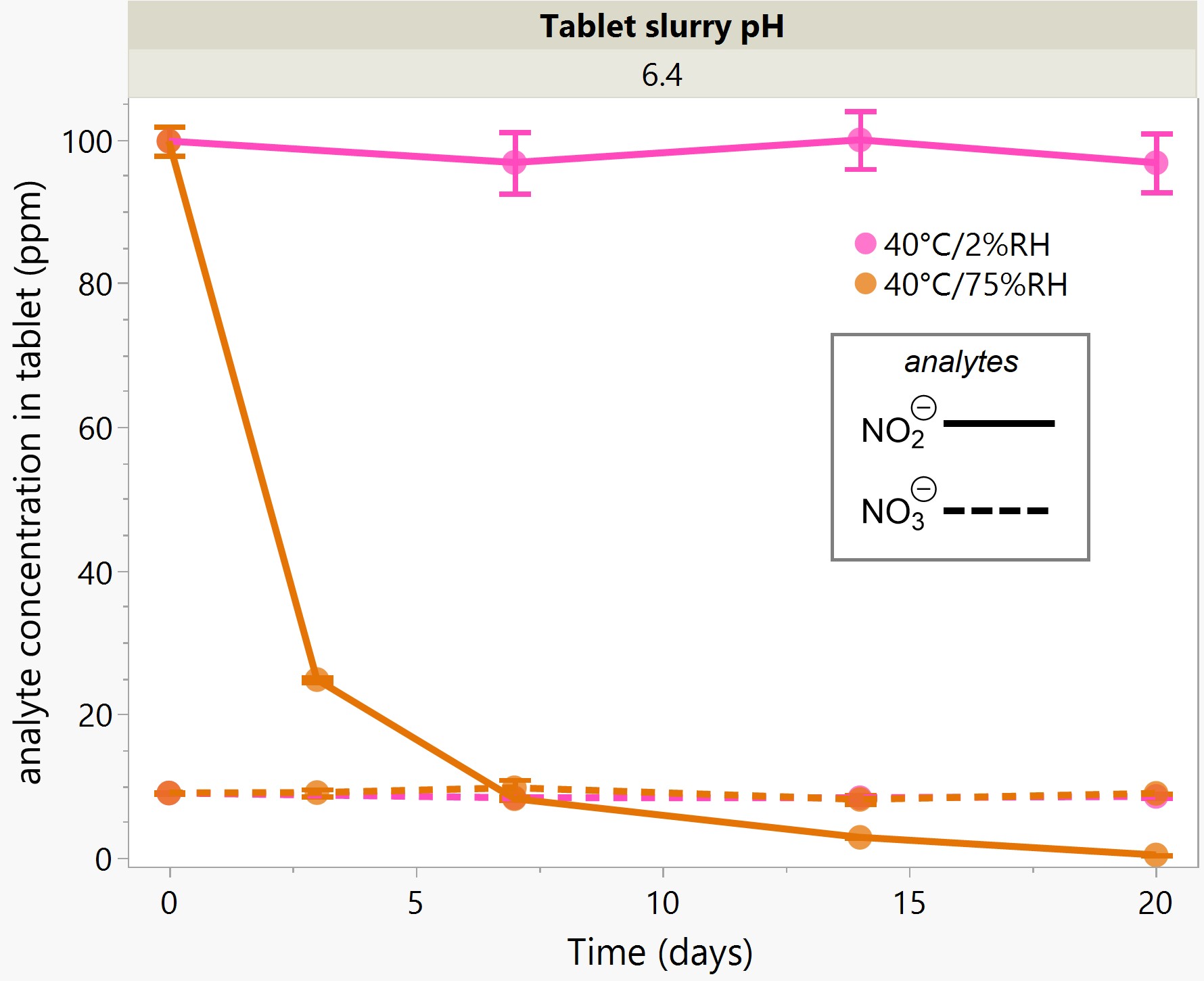
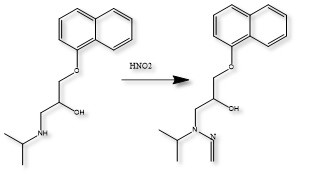
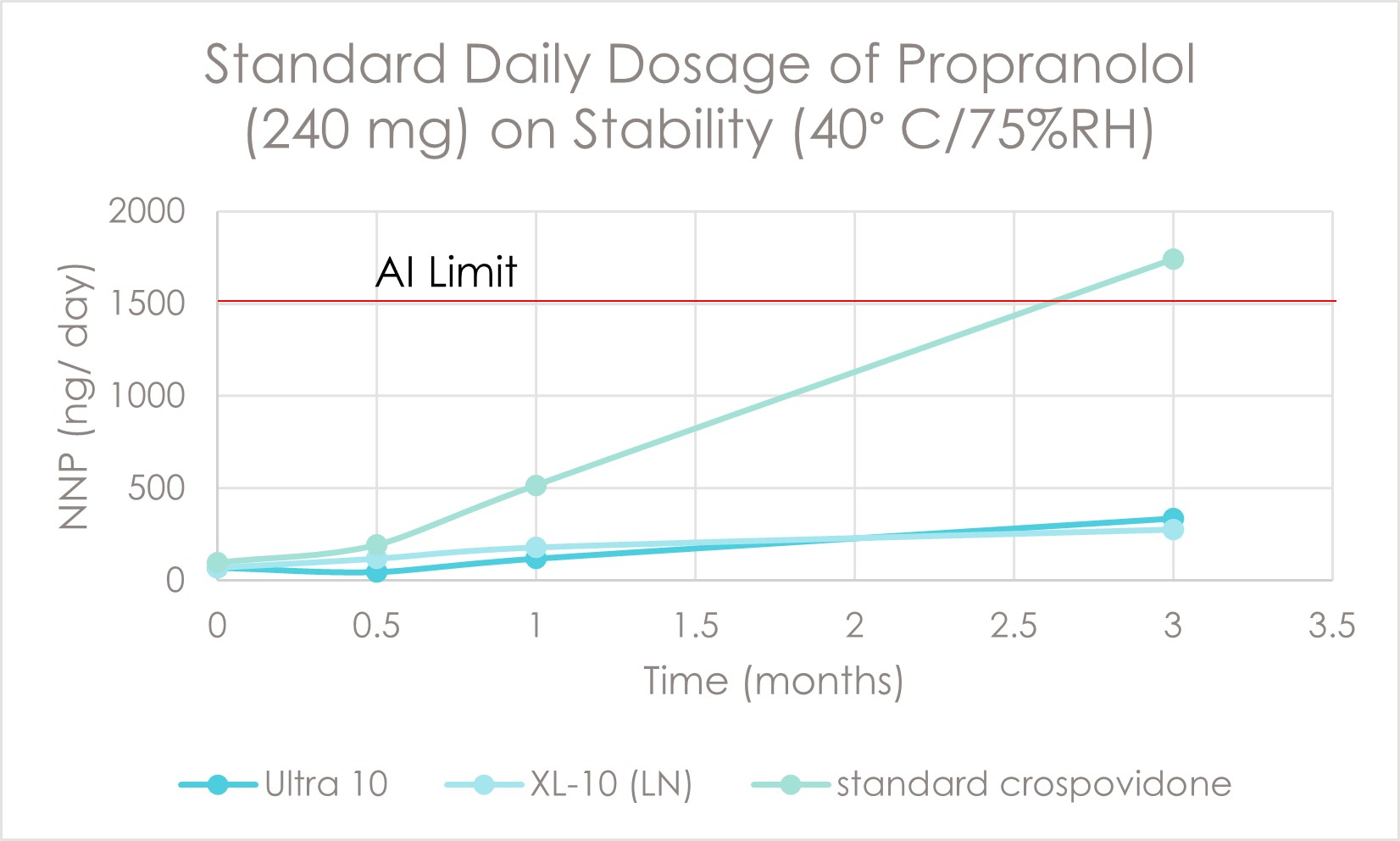
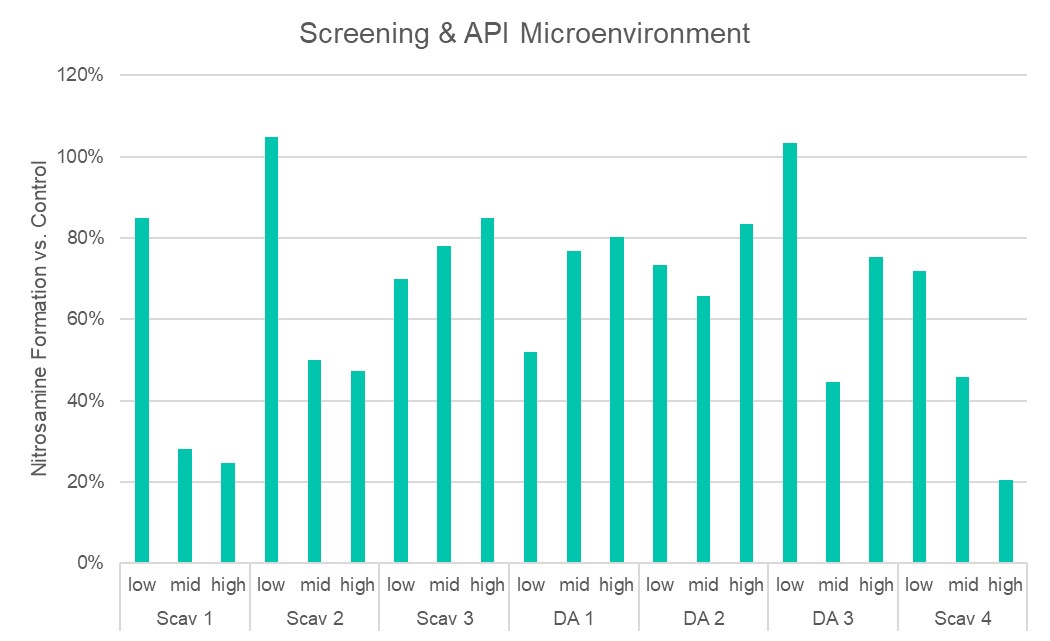
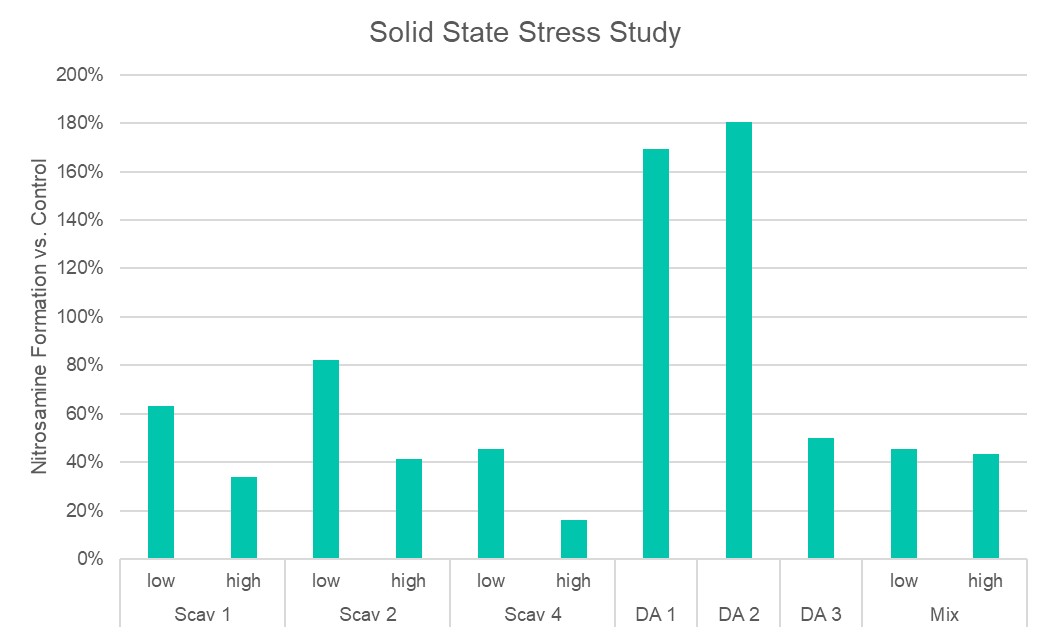
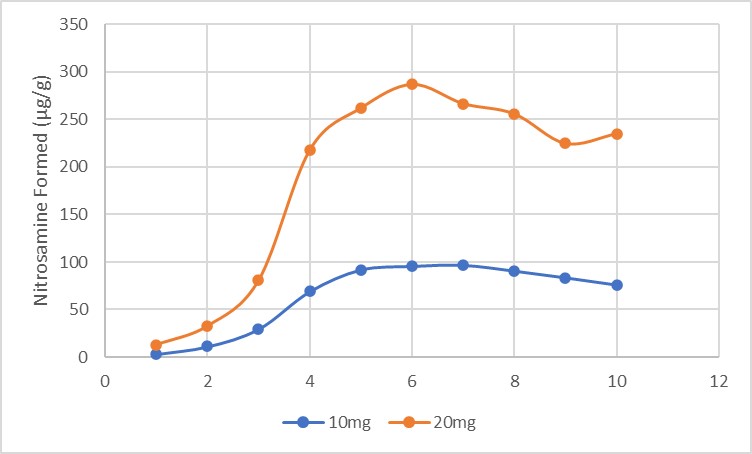
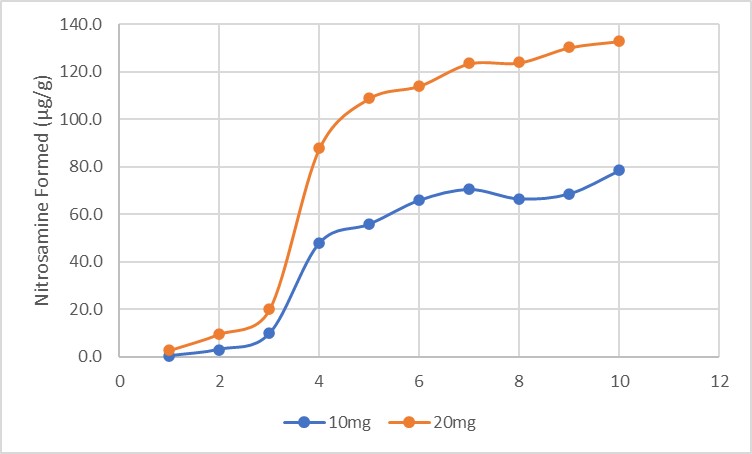
Results: The preliminary data shown that the SSRS, specifically crystal defect, could be generated in multiple model compounds through cryo-milling such as lactose monohydrate. The SEM image indicated the difference between unmilled and 60min cryo-milled lactose monohydrate crystal sample. Our group also proved the T1 for lactose monohydrate was decreased when the milling time increased. This result indicated that the T1 could be used to identify the presence of SSRS (Figure.1). To correlate the T1 with the solid-state degradation rate, our group used Aspirin and Gabapentin as the model compound. Aspirin will degrade to salicylic acid and gabapentin will degrade to Gabapentin lactam in solid-state. Our group measured the stability of milled Aspirin and Gabapentin samples in accelerate condition and T1 for those samples after milling. The data shown that the increasing the milling time could increase the amount of crystal defects in the powder sample, in the other words, the sample contained more SSRS. When the sample contained more SSRS, the sample crystal quality decreased. As a result, the T1 decreased, and the reactivity of the sample increased due to the low crystal quality. As depicted in Figure 2, unmilled Aspirin had a T1 of approximately 57 seconds with 83.7% remaining post-stability test. In contrast, 60 minutes cryo-milled Aspirin sample exhibited a reduced T1 of 14 seconds, with no detectable Aspirin remaining after the stability test. The same phenomena were observed in Aspirin formulation and Gabapentin system. This result indicated that the T1 can serve as a predictive indicator of the degradation rate of drug substances or formulations in the solid state, influenced by the presence of SSRS. Once we established the relationship between T1 and the solid-state degradation rate, our attention shifted to drug substances that degraded into nitrosamines. Ranitidine hydrochloride and 4-phenylpiperidine hydrochloride were chosen as model compounds. Stability testing of ranitidine hydrochloride indicated that NDMA might further degrade under conditions of high humidity. Additionally, NDMA levels in all ranitidine hydrochloride samples stored at various temperatures under sealed conditions was higher than as-received samples. The stability of cryo-milled ranitidine hydrochloride samples under sealed condition at 60 °C shown that the longer cryo-milled sample had worse stability comparing with the unmilled sample. However, ranitidine hydrochloride had a very short T 1 relaxation time due to the methyl groups attached on the structure. These methyl groups can spin and act as relaxation “sinks,” significantly reducing the T1. Our group is currently attempting to run the samples at lower temperatures to decrease the spinning of the methyl groups and thereby increase the T1. We are also exploring the possibility of increasing the T1 by deuteration of the methyl groups to mitigate the “sink” effect. The other model compound in our study, 4-phenylpiperidine and its salt form, 4-phenylpiperidine hydrochloride, is currently undergoing stability testing. We are evaluating multiple sample types, including milled samples, samples with low levels of sodium nitrite impurities, and drug formulations. Our results shown that T1 was decreased when the milling time was increased for both 4-phenylpiperdine and 4-phenylpiperdine hydrochloride samples. (Figure 3)
Conclusion: Based on our preliminary data and experimental results, we conclude the following: SSRS can be produced through the grinding or milling of materials. The quantity of SSRS correlates with both degradation rates and 1H T1 solid-state NMR relaxation times. Ranitidine hydrochloride samples degrade more rapidly the longer they are milled. The degradation product of ranitidine hydrochloride, NDMA, may further degrade under conditions of high humidity. 4-Phenylpiperidine is an effective model compound for correlating degradation due to SSRS with 1H T1 solid-state NMR relaxation times.
References: Wexler, P. and B. D. Anderson (2005). Encyclopedia of toxicology, Academic Press.
Akkaraju, H., Tatia, R., Mane, S. S., Khade, A. B., andDengale, S. J. (2023) A comprehensive review of sources of nitrosamine contamination of pharmaceutical substances and products Regulatory Toxicology and Pharmacology 105355,
López-Rodríguez, R., McManus, J. A., Murphy, N. S., Ott, M. A., & Burns, M. J. (2020). Pathways for N-nitroso compound formation: secondary amines and beyond. Organic Process Research & Development, 24(9), 1558-1585.
Harmon, P. (2023). “Ranitidine: a proposed mechanistic rationale for NDMA formation and a potential control strategy.” Journal of Pharmaceutical Sciences 112(5): 1220-1224.
King, F. J., et al. (2020). “Ranitidine—investigations into the root cause for the presence of N-nitroso-N, N-dimethylamine in ranitidine hydrochloride drug substances and associated drug products.” Organic process research & development 24(12): 2915-2926.
Yokoo, H., et al. (2023). “Advanced Solid-State NMR Analysis of Two Crystal Forms of Ranitidine Hydrochloride: Detection of 1H–14N Intra-/Intermolecular Correlations.” Chemical and Pharmaceutical Bulletin 71(1): 58-63.
United States Pharmacopeial Convention, “United States Pharmacopeia and National Formulary (USP 42-NF 37).” Rockville, MD, U.S.A., pp. 3811–3812, 2019.
Byrn SR, Xu W, Newman AW. Chemical reactivity in solid-state pharmaceuticals: formulation implications. Adv Drug Deliv Rev. 2001 May 16;48(1):115-36. doi: 10.1016/s0169-409x(01)00102-8. PMID: 11325479.
Lubach J. 2007. Applications of Nuclear Magnetic Resonance Spectroscopy to Pharmaceutical Solids. Department of Chemistry, ed.: University of Kansas. p 377.
Dempah, K. E.; Barich, D. H.; Kaushal, A. M.; Zong, Z.; Desai, S. D.; Suryanarayanan R.; Kirsh, L.; Munson, E. J. “Investigating gabapentin polymorphism using solid-state NMR spectroscopy” AAPS PharmSciTech 2013, 14, 19-23.
Jarrells TW, Munson EJ 2022. Comparison of Differential Scanning Calorimetry, Powder X-Ray Diffraction, and Solid-state Nuclear Magnetic Resonance Spectroscopy for Measuring Crystallinity in Amorphous Solid Dispersions - Application to Drug-in-polymer Solubility. J Pharm Sci 111:2765 - 2778.
Moser J, Ashworth IW, Harris L, Hillier MC, Nanda KK, Scrivens G 2023. N-Nitrosamine Formation in Pharmaceutical Solid Drug Products: Experimental Observations. J Pharm Sci 112(5):1255-1267.
He, Y., & Cheng, H. (2016). Degradation of N-nitrosodimethylamine (NDMA) and its precursor dimethylamine (DMA) in mineral micropores induced by microwave irradiation. Water research, 94, 305-314.
Jessica C. Beard and Timothy M. Swager The Journal of Organic Chemistry 2021 86 (3), 2037-2057, OI: 10.1021/acs.joc.0c02774
Lactose monohydrate cryomilled sample SEM image and Solid-state NMR 1H T1 relaxation time.
Gabapentin and Aspirin milled/unmilled sample stability data.
Ranitidine Hydrochloride and 4-phenylpiperdine stability and T1 data.