On Oct 18th Zydus Pharmaceutical submitted a citizen petition to FDA regarding Sitagliptin.
What’s a citizen petition?
A citizen petition is a document submitted to the FDA by an individual or company that requests that FDA issue, amend, or revoke a regulation or order or take or not take some form of administrative action. A citizen petition is different from a general grassroots petition directed at a federal agency because under agency rules, the agency must respond to a citizen petition
The Petitioner requested FDA take the following actions considering the risks presented by Nitrosamine impurities in Sitagliptin containing products:
FDA should withdraw the allowed interim acceptable intake levels of up to 246.7 ng per day as per information published by FDA on August 9, 2022 and set it to the level at 37 ng per day level which presents minimal additional cancer risk when compared to a lifetime of exposure to Nitroso-STG-19.
FDA should update and evaluate the data for Sitagliptin containing drug products FDA Adverse Event Reporting System to determine the severity and occurrence of carcinogenicity.
FDA should ensure that drug product manufacturers update their labeling to include nitrosamine impurities associated carcinogenicity risk as black box warning; and
FDA should ask manufacturer of Sitagliptin containing drug product to develop a Risk Evaluation and Mitigation Strategy designed to reinforce medication use behaviors and actions that support the safe use of Sitagliptin containing medication; and
FDA should ask manufacturer about trend data of all manufactured batches with respect to nitrosamine impurity.
FDA should ask manufacturer to submit and publish the information related to AMES test, mutagenicity potential and chromosomal results submitted to respond assessment report of the European Medicines Agency CHMP and Health Canada
The submission included 3 annexes:
Analytical Method for quantification of NTTP in Sitagliptin
At the end depending who you ask its a business. And this is a clear example of at least partially a commercial decision. Also:
As one supplier is achieving the long term limit, asking to take out not compliance products will gain market share.
Releasing full toxicological data, from where is publicily know at least one company did an Ames and an in-vivo assay seems strange. Not sure if this can be enforced and will make a precedent.
Just taking advantage of the sitagliptin topic… Nitroso-STG-19 is a sitagliptin process related impurity and my question is if the scenario can be extended for linagliptin, I mean, could there be a linagliptin process related impurity with AI looks like Nitroso-STG-19?
I am really curious about the response from the agency and the potential repercussions of this approach. We already saw not the same but a similar approach when Valisure reported on NDMA in Ranitidine.
Supporting the politically correct comment in calling it an interesting strategy, while applauding how every day Industry finds ways to come together on this challenging subject, while balancing with protecting company-specific interests.
They are indeed below (but still studying). I don’t know if the combo metformin and sitagliptin is only taken via combination products, or if (for example due to shortages) doctors would also prescribe separately metformin and sitagliptin? Because then they are apparently proposing tighter limits for NDMA in metformin products, which they also have in their business?
Wasn’t the in vivo TGR assay evaluation recently shared in this forum supporting a 253 ng/day? Interested to see if this influences the ongoing FDA discussions on accepting such studies.
If the temporary limit would have been 492 ng/day, maybe there would have been a better case to ask argumentation from FDA why the temporary limit isn’t removed after 1 year?
Do we think to see also some more explicit LTL comments from FDA?
FDA has reported: “In place of the LTL approach, the FDA implements a flexible approach that allows values higher than the AI as interim limits yet maintaining a 1:100,000 cancer risk.” The recent guidance updates from FDA are still not focusing on LTL too explicitly. But going from 37 to 246.7 ng/day just screams the 6.7 LTL factor doesn’t it (at least in the way how the 178 ng/day from EMA kept having a link with LTL principles)? (Applied in the modified sense we have been seeing in EU since Q&A 22 Rev. 17, but modifying LTL to Q&A 22-like concepts was already discussed in NITWG when FDA opened up possibilities for 246.7 ng/day in this case?). Just wondering if the agency would choose to justifying the 246.7 ng/day with a more explicit reference to LTL concepts. For me it makes sense that FDA doesn’t generally publish an endpoint for the t-AI but would agree that with applicants individually.
On data transparency I would rather challenge regulators in general to evaluate if the scope of their data gathering initiatives (government funded research projects) is aligned with their expectations for applicants (timing-wise and study-focus wise).
This was exactly my fear and I had mentioned this to many of my clients that soon we will start seeing petitions based on how tightly they are able to control nitrosamines and also what I call, “competitive testing”, which means testing other suppliers’ lots’ in the market to make sure they are better. We will start seeing more and more of these and also lawsuits on behalf of people who are taking the drug.
It’s a different topic from those we have been discussing so far. I am surprised that nitrosamines should be dealt with by not only science but also business.
I added the MHLW reports of the NTTP contamination issue.
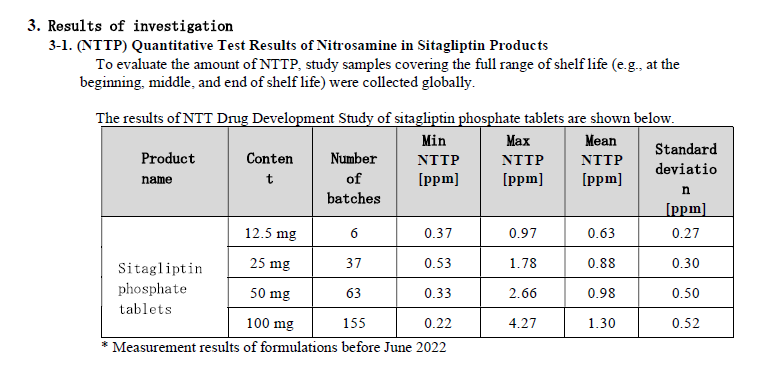
0.37ppm is consistent with 37ng/day. According to the report, root cause investigation and corrective action assessment have been completed. The company seems to have changed the manufacturing process to decrease amounts of NTTP by less than 0.37 ppm.
I understand why the detailed root causes of nitrosamine contamination have not been disclosed in case studies.
Everything is business, as you already know. Generics usually have a very thin line of profit and are trying hard to fight with innovators and also between themselves. I also know a few situations where people are going to try to make immense profit out of this nitrosamine situation. Also, let us be practical about CPCA. We all know that while it is a wonderful hypothesis, it is really not supported by experimental data and will not stand in a court of law.
I would be very surprised if the FDA respond positively to the citizens petition. They have teams of lawyers and the potential for suing the FDA were they to respond positively to a company trying to take advantage of others in this way would be great fun.
When in FDA, I worked a lot with some petitions as the lawyers need scientific advice. These are standard techniques. But it will also make FDA look into their reason for the limit they have proposed. However, I think there are good surrogates of sitagliptin amine with quite robust data and as you said, 37 ng/day is not justified at all. But the concern is that if somebody goes to the court of law and says, unless good experimental data is available, the most conservative limit should be applied, there is a good possibility of that argument winnng and, there may be no turning back. But again, the most conservative limit is 18 ng/day (not 26.4 ng/day if you really look at the available literature), so the company themselves will fail.
Hopefully this not ends in a free for all just for the purpose of profit. I wonder if the petitioners have the 100% of their portfolio evaluated with no issue. Under a scientist view is always a bit puzzling to see some commercial decisions being performed but, well at the end its a business.
The positive part it will give further insights to the public about how limits were derived. I would not say a certain limit adopted by a HA have no evidence at all, but better options may be there. With the caveat that using 18 ng/day or 26,5 ng/day until specific compound evidence is available would put against the wall companies and end in a reformulation that not always would be possible and may not be worth it if a significat higher limit is available later on (not for Sitagliptin I may say as it is a billion dollar molecule).
The uncertain part is how other HA like EMA, HC etc, will see this and again generate region specific discrepancies. Or maybe this would be restricted to the US and its particularities.
This is a case of Science gone legal kind of situation.
Yesterday, there was a discussion on Patient Focused Specifications at PQS 2023.
Am sure the Agency will look into the Scientific, evidence based justifications for this proposal & seek appropriate data in addition to reviewing the supply chain impact.
Collaboration & knowledge sharing is essential to resolve the maze of Nitrosamines.
Thanks @Naiffer_Host for sharing this information with the community.
Surprising and it looks for me this petition have different agenda rather than science based petition as @ASrinivasan mentioned.
However i wonder with following questions and opinions (purely my personal perspective- pardon me if it contradicts others perspectives)
I see nitrosamines issue started with NDMA and NDEA in Valsartan which are generally more potent than NDSRIs- where few judicial courts in USA and Canada already felt that the claim nitrosamines cause really devastating cancer in humans is not backed by sound science- In such context, I am in opinion a petition to be filed based on science and facts rather than asking agency to take certain action.
When an impurity is well evaluated toxicologically with experiments by some pharmaceutical companies and reviewed by distinguished scientific committees although still not approved by leading agencies, i feel at least that should be taken into cognizance before asking agency to take action by petition (Assume petitioner is aware of above data!). It looks NTTP is negative for Ames assay and has BMDL50 of 9.17 mg/kg/day by TGR assay. Despite of that, asking to withdraw interim AI of 246.7 ng/day which is already much lower than expected toxicological limit looks not reasonable.
I believe FDA sets interim limits in balance taking into both science behind safety and also patient need- Ideally FDA would have transparently disclose how they arrived interim limit- Then public will have more confidence. Nevertheless, asking FDA to withdraw current products which are above 37.5 ng/day for NTTP also do not looks rationalistic.
Asking for FDA to issue advisory stating details of contaminated lots to patients and issue quantifying current odds of cancer occurrence in patients exposed to contaminated lots. This step may be appropriate if we have human evidence or at least clue that NTTP caused cancer. Do we have this evidence from patients, who consumed sitagliptin?
This petition also indirectly asks FDA to take one stand either sticking to 37 ng/day or higher AI, it could be interim limit or more than interim limit based on toxicological data.
However, it looks reasonable to propose the following points, but they not only apply for Sitagliptin, it might be applicable to any product with nitrosamine:
Updating and evaluation of FARES
Labelling
REMS and many more…
Hope FDA is doing those…
By and large, any petition/decision by pharmaceutical company or agency should safeguard patient health that is based on science but not on other agenda.
Who will address the drug shortage and patient care if we start withdrawing products with no concrete science!!
I agree. The petition is a wake up call in my opinion. Again, CPCA is a hypothesis. The 26.5 ng/day and 18 ng/day for NDSRIs were hypothesis too. The issue is that the agencies are late in catching up to this game and as usual. We really needed the agencies to do what some big pharma have done, set up the labs and start doing carcinogenicity studies. It is not that tough once you get to it. And again, when modified Ames is negative, we should be able to control the NDSRI in the product at ICH Q3A/B limits unless otherwise proven. Right now, we are being rewarded or punished based on hypothetical conclusions.
If one looks in the net, here is the definition of a hypothesis,
“A hypothesis is an idea or proposition that can be tested by observations or experiments, about the natural world. In order to be considered scientific, hypotheses are subject to scientific evaluation and must be falsifiable, which means that they are worded in such a way that they can be proven to be incorrect.”
However, the CPCA, while the answer to some of our prayers, is a hypothesis not yet supported by tested data. In fact, it would possibly be very tough to prove by proper experiments that all larger nitrosamines should be contained at 1500 ng/day. The team submitting the CP has taken advantage of that fact. They know that the limits are arbitrary and acted on it.
On a different note, a read across can possibly find excellent surrogates for the secondary amine reated to Sitagliptin, some of which were actually negative in robust carc studies done by Dr. Lijinsky.
Just one additional comment and sorry to disagree in some cases but is part of a fruitful discussion. Even though it should be the definitive answer, doing carcinogenic studies is a bit too much and if that is the answer it should take representative compounds not for drug classes but chemical structures or similar. Or Ames positive compounds and done by the agencies to diminish the commercial part. But who decides what compounds and until then what to expect? The carcinogenic studies should be performed until end of lifetime and that would take years. Alternatives like the recent Medicines for Europe paper by Dr. Jonhson are a good example of a different approach.
Nonetheless, molecules that are not longer profitable to invest such big quantities would just simple go dry. Will mid to small sized companies do this? Certainly, not. Will big players share the info? Is better to be proactive in the topic or wait for someone else to come with the solution? As still some hypothesis are not being confirmed or discarded.
Finally, at the end what trial would win in court if it is almost imposible I may say a medicine because of nitrosamines caused cancer when the ingest by other sources is much bigger.
As I read a comment on LinkedIn, this topic should be managed by scientist and not by activists and lawyers.