Hello all,
I am currently working on a new validation (for tablets) and we are now at the robustness testing.
Formation during extraction was observed quite a few times but this time we saw formation only in one of the many batches used. It was by luck that we selected the more critically batch and discovered the formation. Had we chosen the other batch we would not see the formation and therefore generate false positive results.
Our presumption is, that the nitrite levels are quite different in the two batches. But we are currently testing the excipients to back up this claim.
Surprisingly after modification of the method, booth finished products, showed about the same residue of N-Nitroso-API. So it seems, that in solution, the formation is quite different than in the tablets. In our case we found that the big driver is water present in the extraction mixture.
Water was needed, because we wanted a method which is capable to dissolve the tablets, so that titration could be avoided.
Had anyone of you also discovered this ? Or maybe there are some publications available, which describe similar problems ?
If such problems arise really on a batch to batch case, then how would you choose the correct batch for development ?
Regards,
Philipp
Hi @Phil
My samples for Nitrosamine testing conducted outsourcing, and I have observed that results of samples from different stability intervals sent in the same time for analysis have a similar / comparable results that in addition of fluctuation of result along the stability study (ups and downs) no trend of results.
I have requested the outsourcing to investigate about that, and considering using water as a solvent to be in the last step before injection trying to minimize the formation of Nitrosamine due to water.
Investigation results were not justify these fluctuation in results or participation of water in Nitrosamine formation, but the thing which I “feel” the root cause of this issue is error come from test handling by analyst.
I will send the same samples to another lab for analysis and when I get illustration about this issue will post here in this post.
Lack of consistent robustness (fluctuating artifact formation during sample preparation) is not always linked to fluctuating nitrite levels, but for acidic sample preparation it most often is. Transnitrosation or other nitrosating agents or catalysts/boosters less often are for acidic pH sample preparation in my experience.
I have not seen cases where the contamination of nitrosating agents or nitrosation boosters in the reagents for sample preparation caused the problem.
Some have reported seeing absorption of NOx in the solvents for analysis as a problem leading to artifacts though.
In my experience robustness is an issue in many cases and thus many methods focus on nitrite scavenging and/or nucleophile blockage during sample preparation.
You are not sharing a summary of your robustness testing protocol though making it difficult to comment on conclusion drawing or confirm agreement to the robustness study protocol.
Without recovery and repeatability data it is difficult to comment.
Detection differences across methods are not indicative that artifact formation is the only problem (cf. matrix effect or recovery issues and how you deal with those).
I have seen cases where across methods on the same matrix there was a huge difference in artifact formation/tolerability of nitrite contamination, matrix effect and recovery, including examples where for certain methods not only artifact formation during sample preparation was seen, but also significant physical-chemical loss of the nitrosamine (which can be hidden due to artifact formation but revisualised when sample prepping and testing placebo spiked with nitrosamine). At the same time the real nitrosamine level can be different in different batches and be fluctuating over time in the same batch (while the analytical investigation can take weeks if not months), further increasing complexity.
Fluctuations in recovery, low recovery, matrix effects or high RSD on sample can be indicating the reality is more complex than an artifact case.
It is not because method A is suspected to generate artifacts, whereas method B gives lower levels, that the presence of significant artifacts with method A is proven. This depends on the overall comparative suitability method A-B (judging that is difficult with the data provided).
When the API is known or predicted to be low in nitroso-API (often the case) and when studying nitroso-API and not nitroso-API impurities I usually recommend to check in method development robustness by:
- Spiking a placebo (full formula without the API of nitroso-API) with API just prior to sample preparation to reconstitute the drug product formula
- Sample prep and test for nitrosamine for difference spiking scenarios (next to the nitrosamine spikes required for recovery testing): 0-x ppm nitrite, 0 or y wt% of an antioxidant or nitrite scavenger compatible with the method developed (so testing samples without any spike, with only a nitrite spike, with both a nitrite and protectant spike and with only a protectant spike); concurrently different methods can be evaluated (different extraction or different pH (acidic/basic/neutral comparison says a lot about robustness root causes, pH <3 is for many APIs best to block nitrosation by protonation but depends on API pKa and stability).
Preferably the nitrite level as such in the placebo (or the typical nitrite content in the medicine) is known, but the absence of increases upon spiking is often telling enough. (E.g. if the method is robust for 10 ppm extra nitrite compared to the base level in the placebo, knowing the exact nitrite content in the placebo is less critical.)
When requiring nitrite testing though, I would not per se focus on excipient testing for this purpose (cf. NOx absorption risks), but would do drug product nitrite testing (if you do excipient testing: the nitrosating agent or booster can also be coming from reagents during sample preparation).
Limiting the queue time of the samples prepped (or parametrising/monitoring this) can be of importance to evaluate the data.
A more advanced way is to include evaluation of artifacts by spiking with stable isotope labelled API (preferable C or N labelled and not H labelled to get better chromatograms) to check for detection of stable isotope labelled N-nitroso-API. (Though labelled API sometimes can be more expensive compared to getting N-nitroso-API-N15 by nitrosating the API with N15-labelled nitrite for recovery testing and monitoring of matrix effect fluctuations).
If you do the same strategy but on a drug product batch with absence of positivity on sample, I’d also expect some form of subtesting on the same sample (nitrite spike, protectant spike, change of method or sample prep pH (appropriately justified then why other method is more appropriate)).
For N-nitroso-API impurities this is often more complex and requires matrixed robustness (cf. fluctuations in both nitrite and the API impurity should often be evaluated).
Again access to stable isotope labelled API impurity can help.
Thanks for your detailed answers and thoughts
Here some data:
Extraction with MeOH/H20 (2/8)
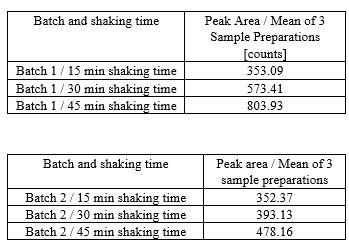
Vit-C was also tested. Unfortunately stability was not given when using Vit-C and the Nitroso-API degraded quite rapidly.
Extraction with only MeOH showed a stable response. We tested the same intervals and had a response of around 350 for booth batches. But then we had to triturate the tablets. Because levels were quite high and it would require a lot of routine testing, this was not optimal.
Recovery was quite good, which values around 95% and RSD<5% across the whole range for booth solvents, when tested with batch 2 and with placebo.
What seemed a little surprising, it that after filtration of the extraction mixture everything was stable even after several days. So increase was only observed when the mixture was in contact with the insoluble components of the tablets. Maybe this was also acting like a catalyst in someway.
Matrix effect was also tested and the influence was not shown.
Everything was extracted using the same LC-MS-System, Solvents, etc. at the same day. So influences should be negligible.
I see your Point with the Robustness testing for different added nitrite levels. This is something we will definitely try for our next validation.
Also the blocking of the amine by protonating it is something we will try if we encounter this problem again. Thank you for ideas.
And for the use of labeled standard, I thought of that myself, but budget is quite tight.
Regards,
Philipp
It is interesting how the hypothesis of “contact with the insoluble components of the tablets” could be supported by the trend observed besides the solvent difference proof. I don’t know the pH reached in the extraction mixture or the nitrosation mechanism to be expected, but it is noteworthy that good linearity for the increase with shaking time is observed, while nitrosation is typically not zero order. Assuming that the levels represented by the peak areas are significant as you suggest (making the more than doubling also significant), I would not per se expect to see a zero order approximation, for example by still being in the linear phase of the curve due to low degree of conversion (which is nonetheless intrinsic for nitrosation of an API) (but I’m biased here not having seen such perfectly linear signs of artifacts formation typically). However, the initial zero order phase approximation can be strengthened if not only the concentration differences in nitrite and API play, but also the accessibility of nitrite from the excipients by dissolution is not perfect (small fraction available for reaction and replenished during the experiment). The linearity might support a not too complex root cause for the artifacts (not multiple mechanisms) (but the fact that artifact add-on can be constant makes it more difficult to notice possibly also in submethods). That zero order is not evident or that accessibility of nitrite plays (while at the same time confirming the suspected nitrosation mechanism) can easily be checked by sample prepping nitrite and API (in the same concentration as with DP sample prep) while parametrising pH (possibly requiring corrects to mimic the situation per real sample preparation).
I’m surely inspired to check more commonly linearity with shaking time in cases where artifact risks are confirmed with nitrite spiking experiments, thank you for this inspiration.

I hope your API is low in nitrosamine so that with a placebo and API spiking experiment you can easily show that also 15 minutes shaking time is not generating artifacts (unless it is obvious that the same values for two methods is not a coincidence). An alternative confirmation can also be a tocopherol spike for the MeOH only sample preparation in case tocopherol is stable with nitroso-API (not all are).
Just some thoughts to possibly (or possibly not) add to this.
The use of methanol only as a solvent does not dissolve all of the other excipients within the formulation, there is still an increase observed with an increase in the shaking time.
The use of 80% water and 20% methanol will presumably be dissolving more of the excipients.
If there is a nitrite rich (relatively) excipient present within the formulation that is only sparing soluble in the methanol, but freely soluble in the water then is this the cause of the reaction then taking place in your sample prep, and the greater the shaking the greater the release of nitrite into solution?
If you change your sample solvent to different ratios of methanol:water then do you see different gradients for the formation, fitting between the two plots shown, as the excipient dissolves? Does it still reach the same maximum value with these lower levels of water, but require a greater shaking time to get there?
If you can “prove” that the additional nitrosamine is coming from the sample preparation, and not from what is inherently present within the tablet prior to the sample preparation then you could start to form a justification for why the methanol only sample preparation gives the “correct” result. Or look at different mechanisms in the sample prep to scavenge the nitrite if that is the cause.
On a side note - it is interesting to note that the Vitamin C breaks down the nitroso-API when added as a scavenger. Vitamin C has been widely postulated as an excipient to add as a scavenger to prevent the nitrosamine formation, but is it preventing the formation in the finished product, or breaking it down again in the sample prep? (And does that ultimately matter, if it means that the nitrosamine is broken down in the stomach when the product is taken?)
Creative thinking on the vitamin C. (I interpreted that the degradation of nitrosoAPI with vitamin C is directly seen, e.g. by testing reference standard or placebo spike with reference standard).
Complexity of ascorbic acid interactions with nitrosating agents and/or nitrosamines in the stomach is probably too high to fully leverage an effect on the real human cancer risk.
So far my understanding would be that also ascorbic acid exogenous nitrosamine formation protection is easier accepted than endogenous formation protection?
(Similarly, I don’t know if there are any examples also on leveraging denitrosation enzyme effects.)
If we accept that nitrite scavenging by ascorbic acid protects in vivo, then the door is open for accepting ascorbic acid nitrosamine detoxification via denitrosation as in vivo protection as well, as both depend on the question whether NO uptake by ascorbic acid can lead to important nitrosative species (leading to nitrosation or net transnitrosation). Formation of NO is only deactivating if no reaction with oxygen takes places to reform nitrosating species (often but not always the case?).
(All this assuming that a nitrosoAPI degradation can be explained by denitrosation, like described for example for N-nitroso tryptophan and N-nitroso melatonin (although not being nitrosamines); doi: 10.1002/chem.200600405)).
There has been some reporting that the inhibitory effect of ascorbic acid for preventing endogenous nitrosation in the stomach can also not be guaranteed in case of a relevant amount of fat in the stomach (doi: 10.1136/gut.2007.128587), possibly because the NO generated from the deactivation by ascorbic acid moves to the lipid phase where it oxidizes to a nitrosating species and interacts with amines.
So far I have failed to proof that in some cases lack of full avoidance of nitrosamine artifact formation during sample preparation in presence of antioxidants like ascorbic acid can be equally explained by oxidation mechanisms or biphasic system effects (cf. partial dissolution problems).
Some Information regarding the Vitamin-C Degradation:
The following things were tested and everything was not stable (no signal was detected anymore after 12h):
Solvent + Vit-C + Nitroso-API in different concentration levels
Placebo + Solvent + Vit-C + Nitroso-API
Finished Product (booth batches) + Solvent + Vit-C + Nitroso-API
Only one amount of Vit-C was tested. In the past we achieved good results with 4% Vit-C in mass added.
Although it looked at first, that the addition was kind of solving the problem (Table shows values obtained for the critical batch):
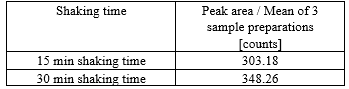
I think maybe formation and Degradation was happening around the same pace. Since a small increase is obtained even with Vit-C present.
Unfortunately we don´t have more data on this, but it would be nice to know, how the signal behave between 30 min and 12h.
Many methods indeed tolerate a wt% level or higher ascorbic acid spike.
I recognise this pattern.
I also dealt with a case where NDSRI artifact formation was seen in a drug product for which the sample dissolution during sample preparation was one of the challenges as well (and the nitrite content was elevated), trying to solve it with a certain antioxidant system led to NDSRI degradation (shown in similar fashion as you describe), whereas also in some experiments nonetheless the NDSRI detected was consistent with other test methods (balance between degradation and formation during testing, occasional good recovery of unlabeled NDSRI but low repeatability). In the end, 7 submethods of 4 key methods were evaluated (varying scavenger use (antioxidants and other) and solvent systems (water-based/extraction-bases vs. water-free), none were fully robust with regards to artifacts (although they could result sometimes in the same detection for the same batch) and varied in the tolerability of nitrite (which was fairly low, considering that many other methods on NDSRIs can tolerate a 20 ppm nitrite spike, whereas here 3-5 ppm nitrite tolerance was difficult). As such a nitrite testing method on drug product was developed to help with the interpretation of NDSRI testing data against the robustness study matrix to evaluate if the limitation of the best available method has an impact on the decision making (unfortunately high daily doses of the medicine in combination with lower CPCA classes increase the criticality of a certain portion of artifacts) or can be tolerated. For validation purposes a nitrite pretest to confirm access to the method and/or a spike with stable isotope labelled NDSRI precursor (to show absence of significant NDSRI-labelled in the chromatogram) can be useful.
When other drug products were tested for the same NDSRI, the same problems did not occur and the matrix-specific method didn’t even need scavenger protection (these drug products didn’t have high nitrite content or the same sample dissolution problems).
Over the years I have learned that when such problems arise, moving early on to the use of (expensive) stable isotope labelled amine for spiking and/or stable isotope labelled NDSRI for spiking (depending on having 1 or 2 issues) sometimes saves money and time compared to keep doing efforts to get similar info in multiple steps.
Each matrix is different, but I agree with a suggestion in a recent workshop that it is likely there have been nitrosamine-related recalls based on artifacts, considering the challenges sometimes to fully proof it are not significantly (both compared to a guided limit and the overall detection) artifacts.