Are you facing significant challenges in your analytical methodology for quantifying nitrosamines?
A collaborative effort involving various regulatory bodies occurred, including: US FDA, Health Canada, ANSM (France), Swissmedic (Switzerland), LGL (Germany), and TGA Laboratories (Australia). This partnership aimed to investigate the consistency of nitrosamine measurements. Each lab contributed its unique analytical methods to assess nitrosamine levels in standardized samples.
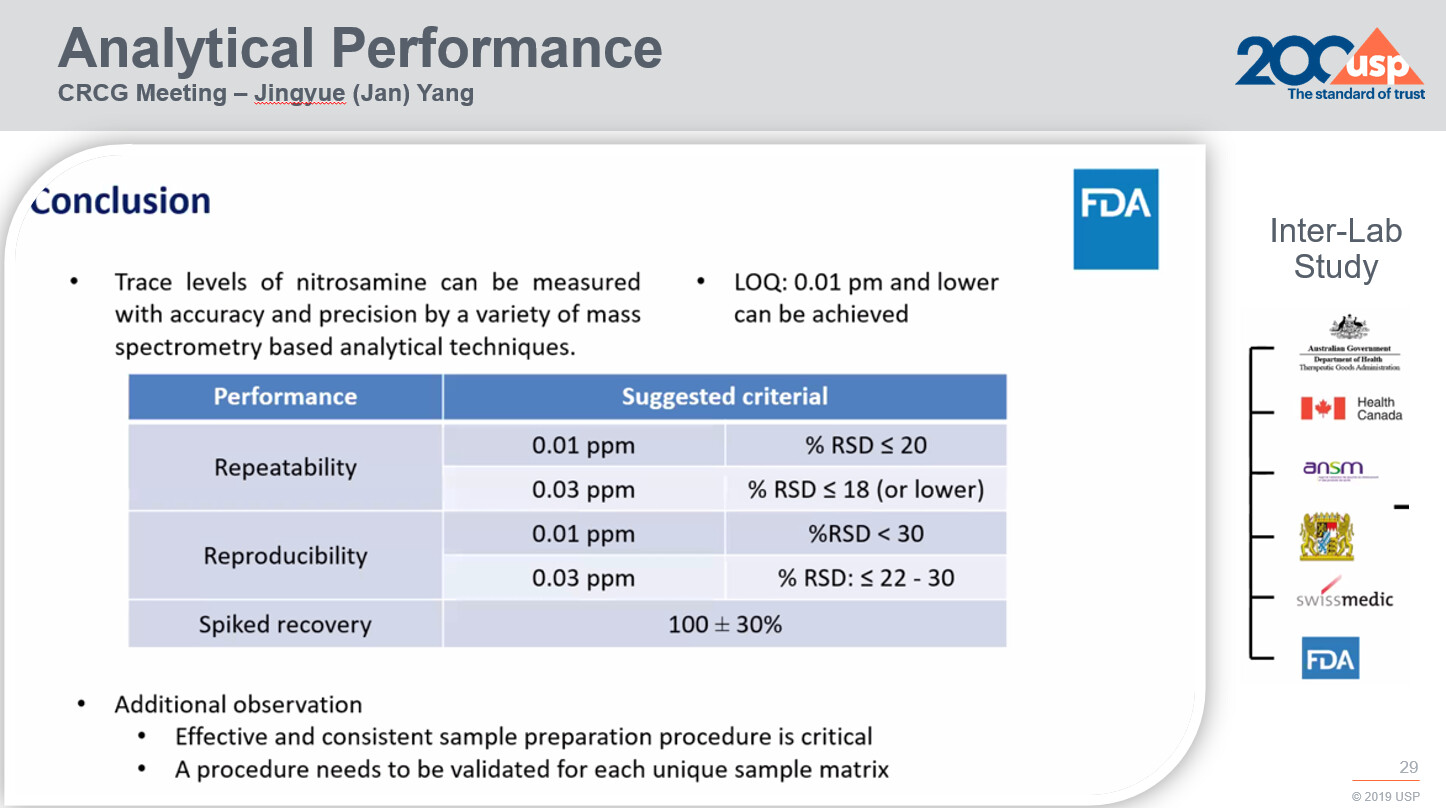
In the paper titled Performance Characteristics of Mass Spectrometry-Based Analytical Procedures for Quantitation of Nitrosamines in Pharmaceuticals: Insights from an Inter-laboratory Study the authors demonstrate their success in accurately and precisely measuring trace-level nitrosamines using diverse analytical techniques. The study also sheds light on how methods relying on mass spectrometry perform in terms of accuracy, consistency, and reproducibility:
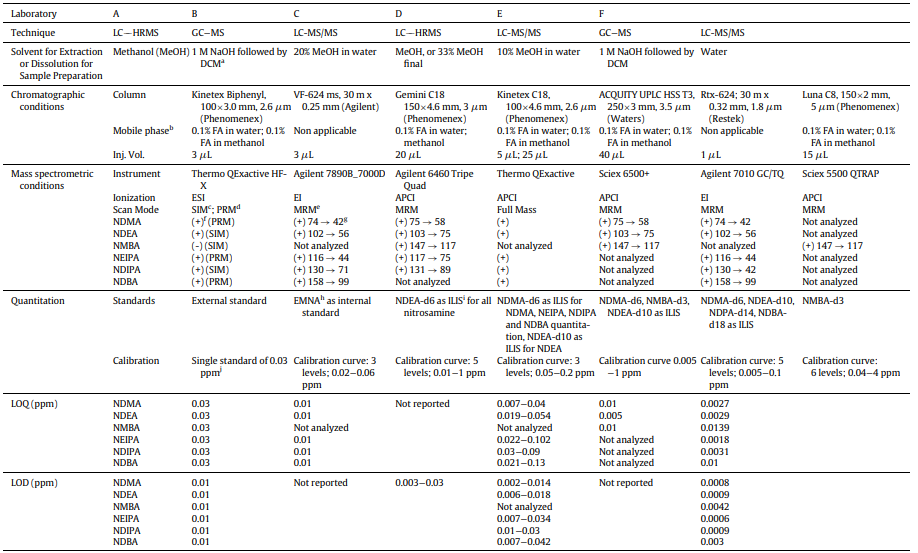
The variations in measurements across labs can be attributed to factors such as:
-
Sample preparation procedure;
-
Matrix effects;
-
Uneven distribution of nitrosamine contents within a sample, and
-
Limited quantitation ranges sample preparation methods.
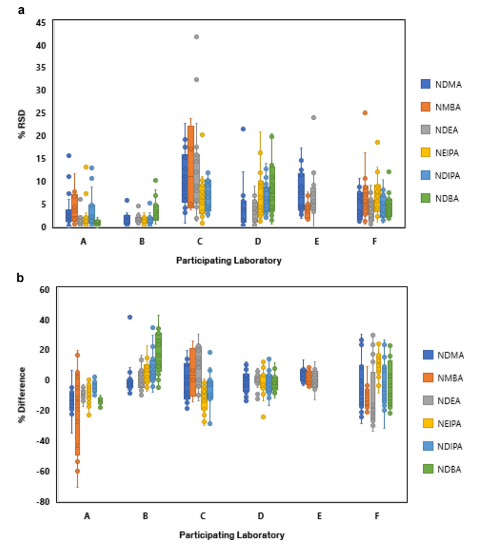
These findings underscore the significance of adapting analytical methods to match the distinctive characteristics of each sample type. These methods should be validated to ensure their suitability within specific measurement limits.
It’s vital to note that an analytical approach proven effective for analyzing nitrosamines in one medical product cannot be automatically considered valid for a different formulation.
Building on these findings, regulatory bodies and stake holders have initiated discussions about the criteria for evaluating MS-based analytical methods. Essential questions revolve around the acceptable level of variance that can be anticipated and considered reasonable for quantification using mass spectrometry.
Do you share similar uncertainties and inquiries about this subject?