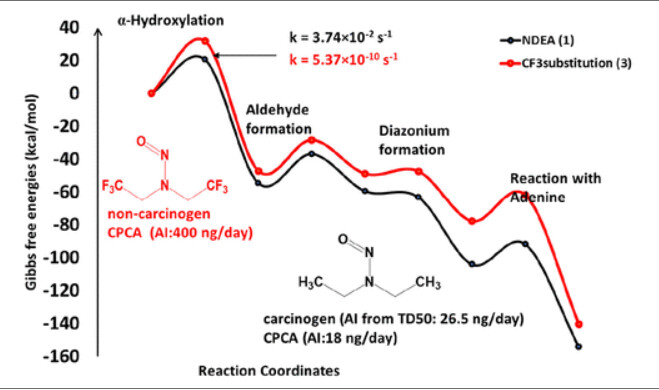
This seems a potentially very interesting piece of research, looking at how the structure of the nitrosamine could impact the reactivity and ultimately the carcinogenicity, linking it to the CPCA.
One especially for those interested in quantum mechanical modelling. I haven’t yet read the full article, need a quiet space for that to try and understand it all.
Abstract:
Nitrosamines are in the cohort of concern (CoC) as determined by regulatory guidance. CoC compounds are considered highly potent carcinogens that need to be limited below the threshold of toxicological concern, 1.5 μg/day. Nitrosamines like NDMA and NDEA require strict control, while novel nitrosamine drug substance-related impurities (NDSRIs) may or may not be characterized as potent carcinogens. A risk assessment based on the structural features of NDSRIs is important in order to predict potency because they lack substance-specific carcinogenicity. Herein, we present a quantum mechanical (QM)-based analysis on structurally diverse sets of nitrosamines to better understand how structure influences the reactivity that could result in carcinogenicity. We describe the potency trend through activation energies corresponding to α-hydroxylation, aldehyde formation, diazonium intermediate formation, reaction with DNA base, and hydrolysis reactions, and other probable metabolic pathways associated with the carcinogenicity of nitrosamines. We evaluated activation energies for selected cases such as N -nitroso pyrrolidines, N -nitroso piperidines, N -nitroso piperazines, N -nitroso morpholines, N -nitroso thiomorpholine, N -methyl nitroso aromatic, fluorine-substituted nitrosamines, and substituted aliphatic nitrosamines. We compare these results to the recent framework of the carcinogenic potency characterization approach (CPCA) proposed by health authorities which is meant to give guidance on acceptable intakes (AI) for NDSRIs lacking substance-specific carcinogenicity data. We show examples where QM modeling and CPCA are aligned and examples where CPCA both underestimates and overestimates the AI. In cases where CPCA predicts high potency for NDSRIs, QM modeling can help better estimate an AI. Our results suggest that a combined mechanistic understanding of α-hydroxylation, aldehyde formation, hydrolysis, and reaction with DNA bases could help identify the structural features that underpin the potency of nitrosamines. We anticipate this work will be a valuable addition to the CPCA and provide a more analytical way to estimate AI for novel NDSRIs.
Calculate C-N bond stretching frequency of the diazonium metabolite of the nitrosamine (measure for the bond dissociation energy and thus diazonium reactivity and thus likelihood of DNA alkylation)
Build correlation with published TD50 data of justifiable analogues
Calculate C-N bond stretching frequency for the NDSRI studied and orient in the spectrum to correlate C-N bond stretching frequency with TD50 to predict TD50 of the studied NDSRI
Abstraction is made of the ease to form the diazonium itself (assumed as easy for the analogues as for the NDSRI itself by approaching the problem based on the diazonium directly)