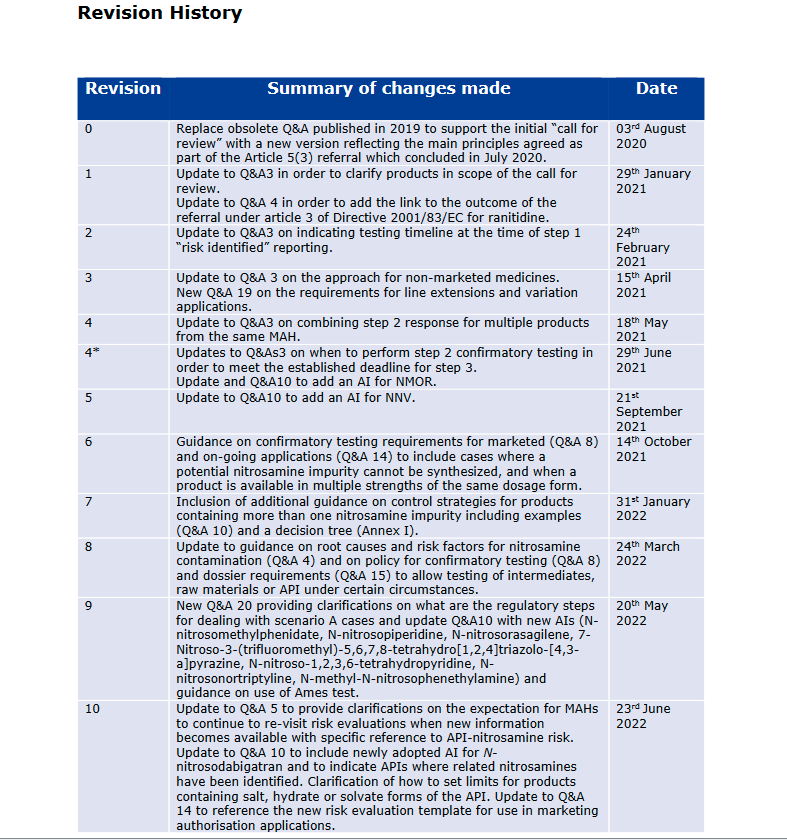
A little detail on the EMA revisions as well:
Q&A document on nitrosamine contamination in February this year, three further updates (Rev. 8 of 24 March, Rev. 9 of 20 May and Rev. 10 of 23 June 2022) have been made in the period of 12 weeks.
The following is a summary of the respective additions in the answer texts to the individual questions:
4. What are the currently identified risk factors for presence of nitrosamines? (Rev. 8)
The risk factors for the formation of nitrosamine impurities were divided into three categories and partly completed by more detailed explanations:
-
Risk factors in the manufacture of the API.
Additions: detailed references to chemical reaction pathways leading to the formation of nitrites as a source of nitrosamine impurities.
-
Risk factors in the manufacture of the finished product
Additions: detailed references to further chemical causes of nitrite formation.
-
Risk factors in relation to GMP aspects.
Additions: Reference to technical literature on the chemistry of nitrosamine formation.
5. What to do if after submission of Step 1 and/or step 2 responses, new information (e.g. related to new potential risk factors or root causes) is identified? (Rev. 10)
In the new wording of the answer, the expectations on the marketing authorisation holders are specified as follows:
- The risk assessment should be re-examined as new information on potential sources of nitrosamine contamination becomes available.
- Questions 4 (risk factors) and 10 (limits) of the Q&A document are to be routinely reviewed for updates.
Particular attention should be paid to potential nitrosamine sources from APIs. A high risk is the simultaneous presence of amines and nitrite traces during formulation of the final product or its storage.
8. How should confirmatory tests be conducted by MAHs and manufacturers? (Rev. 8)
The answer to this question specifies the case where the cause of nitrosamine contamination is only at the stage of manufacture of the API. If this is proven beyond reasonable doubt, confirmatory testing may be limited to the API or intermediates only and need not be extended to the finished product.
The prerequisites for this are:
- No additional risk is identified in the risk assessment of the finished product.
- The testing of an intermediate product does not reveal any additional risk for the subsequent manufacturing steps of the active substance.
- The rationale for the strategy where only the intermediates and the active substance are to be tested is scientifically valid.
The responsibility for the confirmatory testing strategy lies with the marketing authorisation holder. The testing strategy must be justified by the risk assessment of the finished product and documented in the marketing authorisation holder’s pharmaceutical quality system.
10. Which limits apply for nitrosamines in medicinal products? (Rev. 9, Rev. 10)
In the table of nitrosamines, seven additional nitrosamines with the corresponding limits have been added. It is explicitly stated that the authorities do not accept a negative Ames test as sole evidence for the absence of mutagenic properties of a nitrosamine. The fact that some nitrosamines exhibit in vivo mutagenicity despite negative Ames test results puts the significance of this test into perspective and additional testing is required to classify a nitrosamine compound as a Class 5 impurity according to the ICH M7(1) guideline.
In the last update of the answer to this question, N-nitrosodabigatran and the associated limit value were added to the table. The table was extended by an additional column (“Source”) in which the active substances are indicated in each case as a potential source of the corresponding nitrosamines. Furthermore, it is explained in more detail how the limit value must be determined if the active substance is present in the product as a salt, hydrate or in the solvate form.
14. What is the approach for new and ongoing marketing authorisation applications (MAA)? (Rev. 10)
Reference to the new risk assessment template to be used in marketing authorisation applications has been included in the answer text to this question. The template can be found on the CMDh website on nitrosamine impurities.
15. When should a test for nitrosamines be included in the MA dossier? (Rev. 8)
The following clarifications have been introduced in the text of the answer to this question:
-
Standard testing of the finished product for nitrosamines (first paragraph).
Such testing is required when either the cause of the nitrosamine contamination is unclear or it can be demonstrated that the contamination arose during the manufacturing process of the finished product or during its storage. The reference to storage has been newly added to the text.
-
Selection of the control point at the finished product, active substance or intermediate stage (second paragraph).
The requirement for a limit to be set in the finished product specification, even if the control point is not at the finished product stage, has been deleted.
-
Waiver of inclusion of the nitrosamine test in the finished product specification (second section, first sub-item).
If the level of nitrosamines is permanently below 10% of the acceptable intake (AI) related to the active substance or finished product, then the test for nitrosamines does not have to be included in the specification. The reference to the AI of the API is new.
-
Reduced test design according to ICH Q6A (second section, second subsection)
A reduced test for nitrosamines (skip-testing) is accepted if the content of a single nitrosamine is permanently below 30% of the AI related to the active substance or the finished product. The wording “nitrosamine levels” in Rev. 7 of the Q&A document has been replaced by “levels of a single nitrosamine” in Rev. 8.
20. What are the regulatory steps taken by authorities following the identification of an N-nitrosamine exceeding the AI? (Rev. 9)
This question and the corresponding clarifications have been newly included in the document. It describes the regulatory steps taken by authorities for so-called scenario A cases. A scenario A case is when one or more nitrosamines are detected in a finished medicinal product that are above the AI limit or when the sum of all detected nitrosamines exceeds the 1 in a 100,000 lifetime risk.
For scenario A, the following measures are taken:
- A pre-determined authority takes the lead in reviewing the information available and providing a preliminary assessment of the case.
- The Rapid Alert Network (RAN) and Single Point of Contacts (SPOCS) are informed. The criticality of the medicinal product is assessed according to the requirements of the EMA document Criteria for classification of critical medicinal products for human and veterinary use
- The lead authority takes the feedback from the RAN into account when preparing preliminary recommendations.
- The Incident Review Network (IRN) is consulted to facilitate the exchange of information and to assess whether further action needs to be taken.
- If action is recommended in relation to market products, the National Competent Authorities (NCAs) will initiate further action depending on the criticality of the products concerned. In doing so, the lead authority and the NCAs take into account the LTL approach or the use of interim limits for a certain period of time.
In parallel to the update of the EMA/CMDh Q&A document, the corresponding CMDh document containing the practical guidance for holders of national marketing authorisations (including mutual recognition and decentralised marketing authorisations) was also revised.