
@MonazBmysorewala thanks for posting the news…
below the official FDA announcement: FDA works to avoid shortage of sitagliptin following detection of nitrosamine impurity
Does anyone understand or know what Nitroso-STG-19 structure is?
Thanks and welcome Sir!
Hi Villain17, if you don’t know yet, NTTP is
7-Nitroso-3-(trifluoromethyl)-5,6,7,8 tetrahydro[1,2,4]triazolo- [4,3- a]pyrazine
Regards,
Filip
Does anyone know on what terms (study?) did the FDA determined the AI up to 246.7 ng/day?
Any root cause identified for source of this impurity in Sitagliptin. Whether from starting material or degradation .
For combination drug product of Sitagliptin and Metformin , we have NDMA in Metformin with 96 ng/day and Sitaglptin with 37 ng/day as acceptable intake limit. In this case how limit for individual and total impurity can be derived in drug product.
Thanks
Please refer to this earlier post where we discussed the early limits, read-across surrogates, and potential sources
@conudel Can you help us understand the potential read-across on the structure?
@schlinjo1975 Has anybody researched the degradation mechanism of the molecule that could be an indication of the impurity increase during stability?
Thx
Sitagliptin and it’s family of impurities…

Some synthetic routes for impurities 2 , 3 , 5 , 6 , 7 , 9 , and 10 were reported in the scientific and patent literatures, whereas recently identified cyclohexyl impurity 4 was synthesized by Bandichhor and co-authors. Interestingly, synthesis of impurity 9 was reported mainly starting from sitagliptin or late stage intermediates, while only one four-step synthetic procedure from 2,4,5-trifluorophenylacetic acid was reported in a Chinese patent application. Furthermore, two additional steps are needed if 2,4,5-trifluorophenylacetic acid is prepared from readily available 1,2,4,5-tetrafluorobenzene. Similarly, the synthesis of alkene impurity 10 was mentioned only in the patent literature; however, the starting materials were only late stage sitagliptin intermediates or sitagliptin.
Impurity 3 is also formed under acid or basic hydrolysis according to the following degradation study link:
BTW NTTP chemical structure is:
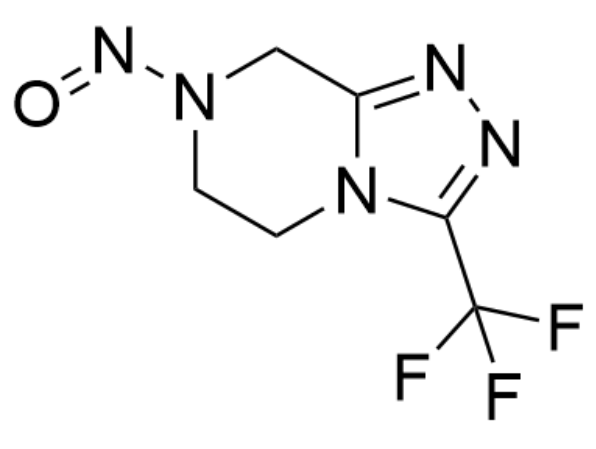
I found an assessment report of the Sitagliptin Accord, which includes the different salt forms from the sitagliptin phosphate monohydrate.
According to the report, three impurities described in the Ph. Eur. in the monograph of sitagliptin phosphate monohydrate are Impurity A (enantiomer), Impurities B, and C. And Impurities B and C are unspecified.
This sitagliptin was considered free from nitrosamine. Please see the following excerpt from the report.
P16.
The Applicant updated the risk assessment to include the discussion on potential nitrosamines likely to be introduced based on the manufacturing process of the drug substance and interactions between the components of the drug substance, excipients and packaging materials. All nitrosamines that could potentially be formed taking into account all parent amine entities (secondary, tertiary and quaternary) that could be present in the components of the drug product (drug substance, excipients, water and packaging materials) have been considered. Based on the additional information provided, overall it was accepted that no risk was identified of the possible presence of nitrosamine impurities in the active substance or the related finished product. This is sufficient and confirmatory testing is not required. The batch results provided by the Applicant (NDMA, NDEA, NDIPA, NEIPA and NDBA in three active substance batches, and NDMA and NDEA in 6 finished product batches for each strength) are considered supportive. The analytical methods used have been adequately described and appropriately validated in accordance with the ICH guidelines.
I’m not sure whether this sitagliptin includes impurity3 or not. I guess it could be identified through risk assessment if contained. And as @Diego_HM pointed out, I’m concerned with acid hydorisis through DP manufacturing or storage. Significant degradation was observed in acidic stress studies of API. What do you think?
@sameer based on the FDA guidance when more than 1 nitrosamine is present…
If more than one of the nitrosamine impurities identified in Table 1 is detected and the total quantity of nitrosamine impurities exceeds 26.5 ng/day (the AI for the most potent nitrosamines) based on the maximum daily dose (MDD), the manufacturer should contact the Agency for evaluation. For drug products with an MDD of less than 880 mg/day, a recommended limit for total nitrosamines of 0.03 ppm is not more than 26.5 ng/day and is considered acceptable. For drug products with an MDD above 880 mg/day, the limit for total nitrosamines should be adjusted so as not to exceed the recommended limit of 26.5 ng/day
Thanks Naiffer for your reply.
Questions and answers for marketing authorisation holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products, 24 March 2022
EMA/409815/2020 Rev.8 (pg 16) provided following options for calculation of total impurities when multiple impurities present.
For option 1 : the AI limit for total N-nitrosamines should be set in ppm/ppb according to the most potent N-nitrosamine present at ≥10% of its AI. The most potent nitrosamine is the one with the lowest AI (see table 1). Limits for individual N-nitrosamines can be defined but are not necessarily needed. However, it should be clearly stated which N-nitrosamines are included in the calculation of total N-nitrosamines.
For option 2: the limits for N-nitrosamines should ensure an overall risk of not more than 1 in 100,000. Different approaches can be employed to achieve this risk requirement:
Fixed approach: fixed AI limits (in ppm/ppb) are set for individual nitrosamines and no limit for total N-nitrosamines is needed. The limit for each N-nitrosamine should be set at a percentage of its AI limit such that the sum of the % AI limits for each specified nitrosamine does not exceed 100%.
Flexible approach: each N-nitrosamine should be specified at its AI limit in ppm/ppb and an additional limit for total N-nitrosamines is required. The calculation for total N-nitrosamines could be written as: 𝑋𝑋
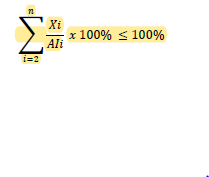
Where Xi is the amount of each single N-nitrosamine i in ppm and AIi is the AI limit of each N-nitrosamine i in ppm.
For each batch, to determine whether the limit for total N-nitrosamines is met, the amount of each N-nitrosamine present (in ppm/ppb) should be converted to a percentage of its respective AI limit. The sum of %AI limits of specified N-nitrosamines should not exceed 100%.
Also provided examples for calculation
Not sure whether this is also accepted by FDA
Thanks & Regards
Sameer K
Dear Sameer,
Thank you for sharing information.
Do you have any insight on LTL approach with examples?
I am aware that FDA generally does not accept LTL approach for NI impurities, however, drug product clinical use is short term treatment.
Thanking you in anticipation.
Regards.
Yes, NTTP impurity is increases during stability. Temperature and humidity plays role for degradation.
TGA also accepts a temporarily acceptable intake of 246 ng per day for NTTP “until 31 May 2023” to avoid a shortage of sitagliptin. Details of the calculation are not found in the following web pages, unfortunately.
Hi All, does anybody have access to the analytical method for the analysis of NTTP in Sitagliptin? I just saw a post on it.
Root cause might be the API ROS but on thermal degradation (about 24 hrs) impurity Content almost showed me double or sometimes triple on LCMS/MS Method.
we have some unexpected observation in Sitagliptin + Metformin HCl extended release tablets. NMDA levels are at 60 degree celsius are lesser compare to samples stored at room temperature. one more interesting observation is that, samples kept at 40 degree celsius 75% RH shows NMDA - not detected.
Generally impurity levels shall be proportional to the temperature and time.
Can anybody helps to understand this observations.