Hi to all,
Yes I agree there is more alignment in the industry on Nitrosamine after it was introduced on Sartan’s family. However there are still a lot of ambiguity with respect to how many Nitrosamine impurities one has monitored in Drug substance and Drug product.
As per guidelines published by USP and EMA there are about 7 impurities listed with a straight limit of 30ppb. The guidelines also suggest to calculate the limit based on Acceptable intake ( AI) & MDD but it doesn’t allow to consider the Less than life time approach ( LTL) factor , at least for the ONCO products where the drug itself is highly potent and the treatment period is almost one or two year in that case why LTL approach is not been considered this needs to have clarity.
Also today industry is confused whether for all category of drugs do they need to evaluate all 7 Nitrosamine impurities or can they consider those impurities which are most probable based on there route of synthesis and paper Risk assessment data.
For an API manufacture there are lot challenges as one as to evaluate his Route of synthesis and also has to consider Starting material ROS which is procured from the vendor.
The usual practice followed is the industry is that they are defining the API in three category -
Category -1 where the secondary amine and Nitrosating agent is a part of process hence it is considered as high risk API wherein fate purge study is been adopted to established a control strategy.
Category -2 where either amine or Nitrosating agent is present in ROS . So in this category Nitrosamine can form due to the Nitrosating agent contributing from water or solvents used in process. Such API’s are considered as at moderate risk so in this case can one evaluate only those impurities which are most likely to form. ( Most likely NDMA and NDEA).
Category -3 In this case neither amine nor Nitrosating agents are present in the overall process hence such API is at low risk. ( In this case can a scientific rational justification is suitable ?)
So to begin with this are few of the concerns to the community.
Hello @Mrunal
Glad to connect with you on this platform. Hope you are doing well.
We employ Flowcharts, check lists, FMEA for Risk Assessments from the CMC perspective.
For Nitrosamines, Risk Assessments also include (Q)SAR assessments from safety & Tox perspective.
This topic is still evolving wherein Regulators & Industry are learning,evolving and engaging in exchanging information. Its an amalgamation of concepts of ICH Q8(R2) & Q9
Hello @Nitesh ,
Glad to connect with you on this forum, I do understand your concerns having worked as an Analytical professional in my career.
Would like to share a few points:
!) The number of Nitrosamines to be monitored would be dependent on the outcome of Risk Assessment of DS & DP.
-
A typical drug substance (DS) Risk Assessment would include evaluation of all stages of the DS synthetic route/process from the key starting materials upto the DS for formation of Nitrosamine Imps considering both process risks and contamination risks. This will require evaluation if amines & nitro compounds & nitrosating agents are employed in the synthetic route during a reaction/work-up/ generated during the process as an impurity or as a reaction by product, Intermediate or an Impurity in the solvents employed for the Process. Use of recovered materials such as solvents, reagents or catalysts also requires evaluation as it can add Nitrosamines as a contaminant.The route of synthesis for key starting materials will also require evaluation if it contains amino or nitro functionality and if a nitrosating agent is employed in the synthesis
-
A typical drug product DP Risk Assessment would include assessment of Risks associated with DS, Excipients containing Nitrosamines/containing amines that have the potential to react with a Nitrosating agent/containing nitrosating agent/ any process specific parameters such as pH, Temp, etc that could lead to Nitrosation, excipient interaction/compatibility issues Container-closure/PM interactions that could be potential sources of Nitrosamines, assessment if Stability study exposures could lead to formation of Nitrosamines, if Cleaning procedures during mfg introduce any potential amines/nitrosating agents that have the potential to form Nitrosamines, cross contamination issues during product changeover
4)The Regulators are engaging with the MAHs in this matter, EMA has a Nitrosamine Implementation Oversight group (NIOG), FDA has the provision of Pre ANDA meetings wherein issues/concerns can be discussed & addressed prior to submissions.
- Following guidances deliberate in depth on this topic.
ICHM7(R1)
EMA’s Q&A, Rev3 published in April 2021:
EMA’s Implementation guideline published in Feb 2021:
EMA’s Assessment report published in June 2020:
Subsequent to issuance of these guidances, Ph Eur published a General chapter 2.5.42 on N- Nitrosamines in Active substances and the Ph Eur monographs for 5 Sartans were revised.
european_pharmacopoeia_n-nitrosamines_in_active_substances.pdf (556.6 KB)
PharmeuropaSartansEN.pdf (708.5 KB)
The FDA guidances, USP chapter links & webinars have already been shared in this group.
The Latam updates were shared by me earlier in this forum.
The Indian Pharmacopoeial Commission is also deliberating on this topic.
This activity involves cross functional teams deliberating on Risk Assesments comprising SMEs from Process Chemistry, Formulators, Analytical Chemists, Safety & Tox experts, Mfg operations, Quality & RA.
Hope this will help.
Thanks.
Hi Dr.Mrunal - I am good, hope same with you, its a long time back we discussed good to see you in this community.
Thanks for clarifying on ONCO products by sharing the reference of EMA / 369136/2020. at pg.79 under point no.6 it is mentioned ONCO products as exceptions as intended for advance cancer treatment hence N-Nitrosamine impurities should be controlled according to ICH Q3A (R2) and ICH Q3B (R2) as per the reference of ICH S9 guidelines Q&A , pg.no.13
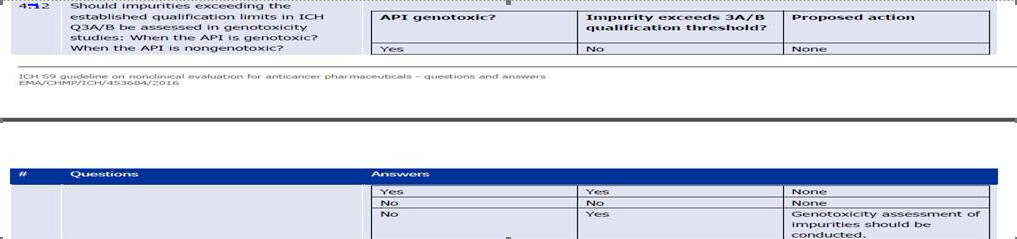
However the concern here is the table does not talk specially about N-Nitrosamine impurities it only says Genotoxic hence regulators at present are not accepting this rational and demands the complete assessment of Nitrosamine as per EMA /369136/2020 also they don’t allow us to consider LTL approach while calculating the limits therefore this needs more transparency in the said guidelines so as to help industry to over come this challenge.
At present our approach to calculate the limit is based on Acceptable intake ( AI) and MDD for all therapeutic category of drugs.
@Nitesh @Mrunal @DAB It’s my understanding that LTL approach has been rule out from N-nitrosamine because the impurity outperform the DNA repair mechanism. However, there are couple great publications that present the framework to adopt this LTL framework. @SusanFelter @jbercu are both authors on the papers. Maybe they can provide some perspective on that, and what we need to give that step forward?.
@Naiffer_Host
The scope of ICH M7 states “This guideline does not apply to drug substances and drug products intended for advanced cancer indications as defined in the scope of ICH S9 (Ref. 4).” Hence LTL approach is ruled out for Onco products.
EMAs Q&A published in April 2021 clearly states "For products intended for advanced cancer only as defined in the scope of the ICH S9 guideline, N-nitrosamine impurities should be controlled according to ICH Q3A(R2) and ICH Q3B(R2) guidelines, as specified in the Q&A document to ICH S9 guideline.
This provides clarity for evaluating limits for Onco products to the mfrs.
Inputs from SMEs in the Forum would be appreciated.
@Nitesh @DAB @Naiffer: This question is often asked by the API and DP manufacturers in nitrosamine webinars/ workshops. The answers have always been ‘These AI limits are not applicable to ONCO products’. Let us hear from other experts too.
Because ICH M7R1 scope is not applicable to Onco products, the Tox & Safety experts can comment on the reasoning but the EMA mandate is to employ Q3A &Q3Bfor Imps in Onco products
Hello @Dr.Archana
Regret for late reply , Yeah absolutely the information is very helpful.
Can you elaborate which analytical technique’s is been followed to demonstrate the Nitrosamines.
The guidelines usually talks about LCMS , GCMS , HRMS techniques however each has its own limitations. Currently what I understand every pharma industry is facing challenges to develop sensitive & reproducible method for API / DP.
Hi @Nitesh ,
The testing for Nitrosamines can be broadly classified into separation, detection, identification & quantification .
The Analytical challenges are being addressed by the Industry & Regulators.
HR MS is being extensively employed for detection purposes because of the sensitivity levels required. The choice of chromatography is dependent on the sample matrix under evaluation, volatile nitrosamines by GC & nonvolatiles by LC. Choice of an appropriate stationary phase also requires attention as these moieties are polar in nature.
As far as DS are concerned, there are published methods available as shared earlier .
Regarding DPs, the onus is on the manufacturer to develop & validate methods for the detection of Nitrosamines in the intended dosage form
Hi Dr.Archana ,
Yeah I too agree HRMS is the most preferred analytical tool which is been adopted in various labs however the challenges of nitrosamine analysis in pharmaceuticals selectivity, and compliance all in the light of obtaining reliable and timely results; high sensitivity must be achieved to fulfill the regulation requirements, and selectivity is also critical to avoid false positive non-compliant results.
Even FDA has published several analytical methods that may be considered when determining nitrosamine content in active pharmaceutical ingredient (API) or finished pharmaceutical product (FPP).These methods include both liquid chromatography (LC) and gas chromatography (GC) coupled with mass spectrometry (MS) or high resolution accurate mass (HRAM) mass spectrometry. LC-MS methods have been developed to cover a wider range of analytes which are not amenable by GC-MS methods. In particular, GC-MS methods cannot directly detect N-nitroso-N-methyl-4-aminobutyric acid (NMBA) and so sample derivatization is required, increasing sample preparation time and efforts. Moreover, LC-MS methods offer a suitable solution when ranitidine is tested for N-nitrosamine impurities. However, the FDA reported that possible degradation effects can occur when ranitidine is stored or analyzed at high temperatures resulting in subsequent formation of NDMA. For the determination of the most volatile nitrosamine impurities, headspace sampling technique is preferred as no sample preparation is required. One of the advantages of using this technique is that it removes the complexity of the matrix while improving the selectivity
for the compounds of interest and reducing the risk of false positive results.
Hence based on matrix effect and which Nitrosamine impurity is to be monitored accordingly the technique may vary.
There is general ambiguity in pharma whether Nitrosamine impurity monitoring should be a part of your final release API / DS specification or it can be additional test off-line data has to be generated this needs clarity.
Hi Dr Nitesh
There is no ambiguity from the Regulators perspective wrt strategy to be adopted wrt Nitrosamine Risk Assessments, testing and control. Evaluation of Imps of Genotoxic potential is a routine exercise while working on NCE candidates, it’s perhaps new to Generics . The Regulatory agencies have conducted several webinar sessions on this topic and are constantly engaged in discussion with the Industry . Most of these sessions are available online as recordings.You can also engage in communication with them for product specific queries … If the process or product doesnot show any potential risk of formation of Nitrosamines then there’s no need to test, however if the Risk Assessment does indicate formation of Nitrosamines then it can’t be treated as additional test off line data.
From tox point of view, ICH Q3 A and B talk of elemental impurities in API and finished product. So final outcome is PDE or ADE. ICH M7 talks about AI. It also does not applies to biological and biotechnology products. So, no matter once the products may have starting material as carcinogenic or mutagenic… it is all about dose decides toxicity. So, as per all learned members, PDE suffuses the best purpose… lifetime exposure without causing harm…be as COV or impurity.
LTL I don’t understand. It’s a fixed dose and limit-based, irrespective of toxicity or severity patterns of an API or impurity. But considering LTL for NA, there is no verdict from regulatory.
Hi ,
Yes the LTL approach is not considered for Nitrosamine unlike GTI’s , this is as per EMA /369136/20202 guidelines published on 25th Jun 2020 which states that – As a precautionary measure, CHMP does not recommend to generally applying the LTL approach to N-nitrosamine impurities. The concept of adjusting acceptable daily intake levels for mutagenic impurities for the expected (less than lifetime) duration of use is outlined in ICH M7(R1) was supported by SWP (however only in combination with the ALARP principle) but was rejected by the ad-hoc expert group because of the potential risk of exceeding individual DNA repair capacity with exposure to high acute nitrosamine doses that may result from implementation of the LTL approach, especially for medicinal products with only short-term use. I believe this is the verdict which regulatory has considered for not applying LTL approach.
I came across the final report on FDA’s health risk assessment Workshop, this is what panelist shared:
- Given that humans are exposed endogenously and exogenously to nitrosamines, the differences in DNA repair capacity among humans as well as among animal species, and the less than ideal quality of nitrosamine carcinogenicity studies, the expert panelists were asked if an in vivo exposure level that can define low vs. high risk for cancer could be identified and whether a NOEL dose could be established.
- Several published studies have shown a clear and abrupt transition of the dose-response to no toxicity, whereas other nitrosamines showed a gradual change with a curvilinear dose response and a sigmoidal curve at low doses. It is important to determine the dose rate, i.e., the interval between doses, and how it affects DNA repair.
- Earlier studies showed that the cancer rate is independent on age. Indeed, when NDEA at the same doses was administered to animal species of different life expectancies, all animals developed tumors at the same rate and time. Issues over the applicability of the LTL approach (ICH M7) to nitrosamines include the reliability of the models for extrapolating from long to short durations, model sensitivity, and the shape of the dose-response curve (nonlinear vs. threshold). These are important points to consider when discussing the question of LTL. The expert panelists did not consider deriving a NOEL for nitrosamines to be appropriate, and it is not accepted for genotoxic carcinogens.
A lot of my learning in this area has been from @jbercu @SusanFelter @conudel @David @kpcross work, publications and presentations. Really interested to get their perspective in all these.
Since this FDA workshop in March a new paper by @jbercu Joel Bercu on LTL and NDEA has been published. It can be found here:
ANVISA now considers LTL limits for nitrosamines acceptable based upon this paper.
Other regulators are looking again at this issue. @kenyom01 @David @fernandaw
can u please share the reference for this? it is a wonderful response to LTL approach.
Great Share Naiffer @Naiffer_Host , this paper goes Indepth into the OELs & LTL limits.
Attached is the PDF of this document that I used for reading, its worth a re read
LTL.pdf (651.0 KB)